

- No Results
- Global
-
Australia

-
Austria

-
Azerbaijan

-
Brazil

-
Belgium

-
Canada

-
Chile

-
China

-
Costa Rica

-
Croatia

-
Czech Republic

-
Denmark

-
ESTONIA

-
Finland

-
France

-
Germany

-
Hong Kong

-
Hungary

-
India

-
Italy

-
Ireland

-
Japan

-
Korea

-
Latvia

-
Lithuania

-
Malaysia

-
Mexico

-
Morocco

-
Netherlands

-
New Zealand

-
Norway

-
Philippines

-
Poland

-
Portugal

-
Romania

-
Singapore

-
Slovakia

-
Slovenia

-
Spain

-
Sweden

-
Switzerland

-
Taiwan

-
Turkey

-
United Kingdom

-
UNITED ARAB EMIRATES

-
United States

-
Vietnam

A New Valuation: How Regulatory is Becoming an Increasingly Valuable Asset
Contact us
Hal Stowe, Senior Manager, Regulatory Intelligence
Have questions for our author? Schedule a meeting.
Regulatory’s New Valuation — Competitive Infrastructure
The companies defining the next decade of medical device innovation are being selected right now, and the filter is not technology — it's regulatory sophistication. The era where technological innovation and clinical impact were sole selection criteria for investors is behind us. Today, Medtech innovation is increasingly defined by risk; it’s preemption, minimization, and strategic integration – all while shifting the burden of risk to sponsors at the earliest of development phases. In this compressed capital environment, bolstered by an FDA which prefers policy from the podium over industry engagement, regulatory strategy and intelligence have moved from supporting function to a primary value driver.
This demand for investment in regulatory strategy will ring true for founders who have built before, and investors who have seen what regulatory missteps actually cost their cap tables. The role of regulatory intelligence and strategy is no longer solely to usher through premarket submissions or urgently respond FDA’s identified deficiencies. The reality is that sponsors must build companies that support commercial launch; not incubators curating intellectual property for acquisition. For this reason, regulatory must not be treated as a star soloist – but as the orchestra conductor turning data and policy from white noise to a symphony, through strategy and market intelligence.
This piece will not navigate general considerations; or provide 510(k) explainers. It will assess the micro- and macroeconomics of the current regulatory and capital environment, providing insight into strategic consequences for sponsors and investors.
Regulatory Policy - No Longer a Predictable Variable
The seismic shifts in FDA policy and personnel are undeniable, with their effects yet to be fully realized. Across CDRH, CBER, and CDER, daily operations are encumbered by the effects of staffing attrition (reductions in force), mounting political pressures, and an evolving enforcement posture unprecedented to industry veterans.
- Staff Attrition: 22% of CDRH Staff (approx. 250 individuals)1
- 18% of FDA (approx. 3,500 individuals)
- Evolving Postures: Political Pressures, Regulatory Priorities, & Enforcement Strategies
- Policy from the Podium: FDA’s use of press releases, speeches, and podcasts to signal policy shifts.
- HHS/FDA Deregulatory Agenda (Executive Order 14192)
- Evolving use of Level II Guidance
- CDRH’s Ambitious FY2026 Policy Agenda
On their own, each of these variables represent casual conditions for supply-side policy shocks. When deployed in concert, their impacts manifest practical consequences for Medtech startups. Review timelines are less predictable, guidance documents carry less certainty (and potentially greater weight), while the informal institutional knowledge relied on by experienced regulatory teams rapidly erodes.
These conditions have unveiled a growing asymmetry; diverging tolerance of regulatory risk between large-cap and startup Medtech firms. This tolerance gap, best depicted in a K-Shaped Recovery Curve, demonstrates the increasingly growing gap in ability to absorb (tolerate) regulatory risk associated with a single (non-specified) decision. This divergence in risk tolerance occurs at the micro-scale (per stimuli), and the macro-scale (firm wide) – with absorption response constant with respect to company size.
 Displayed is the ability for large Medtech firms to absorb regulatory uncertainty into their regulatory infrastructure, while indicating the negative impacts on tolerance as startups absorb risk into their runway. This reduced absorption ability reduces error margins for subsequent regulatory actions or validation tests.
Displayed is the ability for large Medtech firms to absorb regulatory uncertainty into their regulatory infrastructure, while indicating the negative impacts on tolerance as startups absorb risk into their runway. This reduced absorption ability reduces error margins for subsequent regulatory actions or validation tests.
The stem represents the common conditions of regulatory risk shared by both companies, prior to the divergence point. The divergence point marks the threshold at which structural differences between large medtech companies and startups become consequential — where regulatory expertise, organizational resources, and financial resilience begin to determine whether a company can sustain or absorb increasing regulatory pressure.
Below (prior) this point, regulatory challenges are similar. Above it, the gap compounds, with large medtech leveraging advantages to press forward while startups face mounting strain. The shaded gap between the two arms visually reinforces that the tolerance differential widens as risk grows — regardless of where on the risk spectrum you look. The impacts of evolving regulatory risk on that gap are depicted below.
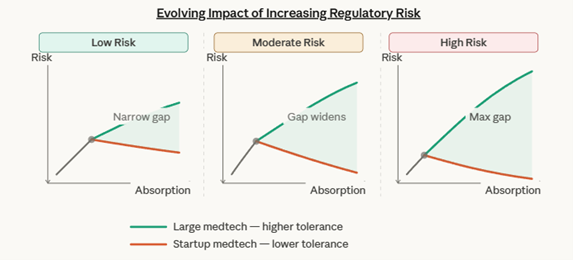
These figures2 provide visual validation of the value contributed through regulatory sophistication. For startups, early investment in regulatory strategy prolongs the divergence point – and may narrow the gap of absorbing regulatory risk. For investors, they reiterate regulatory strategy’s contribution to durable value – in assessing a startups ability to absorb relative regulatory risk.
For early-stage companies, and their investors, risk compounds quickly – and insulative margins-of-error quickly dissipate. A misread-on device classification or submission pathway in year one does not just delay milestones. It can reset capital raise, strain syndicate relationships, and in some cases undermine commercial viability altogether. For late-stage companies, these risks do not dissipate – instead shifting to exit. From experience, regulatory overhangs (and ability to resolve them) become one of the most common causes of a strategics’ re-price, or from abandonment of late-stage deals.
This asymmetry underscores three key items for startups. One, material gains from positioning regulatory as program conductor. Two, a startups’ need to utilize regulatory strategy and intelligence as a scalpel. And three, the close correlation of compressed capital and minimized margin-of-error for regulatory risk. Each of which necessitates reorienting the role of regulatory strategy and intelligence from concept through commercial.
How the Capital Stack Is Responding
Venture Capital: Diligence Has Moved Upstream
The post-pandemic reset to Venture Capital has done more than tighten check allocation. It has fundamentally restructured acceptable risk profiles – and what VC is willing to price into deals. Regulatory risk is a, if not the, leading variable in that recalculation. For venture partners, regulatory risk has moved up to the top of the diligence hierarchy. Once an assumption informally underwritten into a capital raise; the reality of increased product development costs, higher regulatory scrutiny, and longer (7-10 year) exit timelines have all but eliminated that practice. It is now a hard diligence variable.
In response, sophisticated early-stage investors have recalculated the requisites to taking a lead position. Sponsors now must bring to the table a credible (if not validated) regulatory pathway, evidence of FDA engagement strategy (i.e., informational meeting or Q-submission), and a preliminary hypothesis for reimbursement (at minimum). My message to founders (clients or otherwise) is simple. If you cannot speak fluently to your regulatory strategy, you are not fundable to the level needed.
A second message to both founders and investors; proficiency does not equal fluency. When speaking with investors, founders’ familiarity with regulatory strategy must exceed even a proficient understanding of processes and milestones.
Founders must demonstrate fluency in their regulatory strategy, conveying the ‘why’ and ‘how’ precisely, and accurately. They must demonstrate value in their ability to navigate complexities and nuance, and in their ability to account for future obstacles – including negotiating with regulators on proposed indications for use, clinical trial design, or contents of device labeling. For these companies, the ability to identify and communicate the micro (device specific) into the macro context (industry and agency), position founders for early-stage funding success.
The downstream effects of this investment are evident. Startups investing in regulatory intelligence early are compressing their Series A and B timelines — and investors know it.
Strategic Capital: The Bar Has Permanently Raised
There is an ultimate exit path for most Medtech startups – acquisition via strategic investor, and in some cases IPO. For these strategic investors we have seen a shift in diligence posture. Regulatory due diligence is being conducted in a manner mirroring premarket submission review – that’s PMA, not 510(k).
To a previous point, late-stage investor interest aligns with commercial-ready companies – those built with the intent to stand alone, but positioned for acquisition. There is now intense investor focus on QMS maturity, complaint handling hygiene, post-market surveillance architecture, and clinical data integrity. Each of these functions are requisite in standing-up that commercial-ready company, and all of which are now evaluated at the term sheet, not post-LOI.
For late-stage companies, regulatory readiness is now increasingly a differentiator in a premium exit and a distressed one. The strategics’ calculus has changed – no longer buying just an innovative technology. They are buying portfolio- expanding assets, acutely aligned with 5 to10-year commercial plans. Critically, they are buying a regulatory position for that asset; one constructed to facilitate continued innovation and commercial growth through expanded indication, patient population, or integration with other platforms. Investors are now buying regulatory - and pricing accordingly.
Early Validation Demand - A New Strategic Moat
In Medtech product development circles, the concept of a minimum valuable product (MVP) has been a source of thorough debate and discussion for the past few years. It is an incredibly salient topic, as over-engineering in early-stage device development present diminished returns on investment through substantially higher costs, and minimal additional patient benefit. For early-stage startups, early alignment on characteristics of that MVP is critical, as it represents the ideal design iteration for early feasibility and validation. This initial alignment on MVP allows for founders to shift focus and resources, to product validation – the most critical variable to early-stage investors. To efficiently assess and execute early validation, sponsors must development a plan for Minimum Viable Validation (MVV) – the strategic early-stage testing requisite to prove their device’s clinical and economic utility, prior to heavy investment.
For sponsors addressing investor demand for early validation of safety, efficacy, user feedback, and regulatory strategy – determining minimum viable validation (MVV) is critical metric for creating durable value. I will note here that the suggestion made here is not to cut corners in data generation. It is that early-stage companies often engage in early testing without FDA engagement to validate testing strategy, and execution architecture. For companies not utilizing regulatory intelligence at this phase – the result is often superfluous or misaligned data. Data that does not address key investor concerns and may even raise additional concerns of leadership and resource management.
I am always a proponent of early validation. For startups’, the ability to confirm early-stage bench performance, receive formal FDA feedback on premarket submission path, and develop an early prospective clinical evidence strategy are the new signals of a company that understands how to win — not just how to build. It signals organizational and technical sophistication to investors whilst demonstrating a company’s investment itself – identifying and mitigating potential barriers at inception.
For early-stage teams, even limited clinical and non-clinical data early-stage feasibility data, when framed correctly against a regulatory and reimbursement strategy, can dramatically shift the investor conversation. From experience, this ability to tactfully contextualize feasibility data is a variable which shifts investor conversations beyond “show me the technology", to "show me the path”. The ability for founders to utilize regulatory to synthesize, streamline, and advance cross-functional operations (at early stage) is further supportive of investor preference for risk minimization.
When utilized from early concept and validation stages, regulatory intelligence shapes product architecture. A startups’ ability to bring regulatory strategy into developing design controls, framing intended use and indication for use, and the selection of predicate/refence device(s) demonstrates ability to use regulatory offensively and defensively– engineering competitive advantage into the device itself.
There is more to developing the proverbial moat than including regulatory affairs in materials selection, bench testing strategy, and prospective study design. Successful technical validation (bench or animal data) is a necessary condition for durable value creation but is not (generally) sufficient. Early-stage validation must include strategic dialogue with the FDA. Not only to receive necessary feedback (or alignment) on strategy, but to build critical institutional credibility and rapport with regulators. Avoiding the pre-submission process is not a viable strategy for early-stage startups, nor is it an enticing risk profile for investors. Sophisticated pre-submission strategy is another signal of organizational maturity, and a company’s investment in the foundation of their own success.
At early-stage, most founding teams are technical experts – exceptional at navigating and explaining the science and clinical problem. They are, however, often systematically underinvested in regulatory intelligence as an organizational capability. I would note this characterization is changing (positively), yet still greatly outpaced by demand for regulatory strategy.
For startup founders strategically investing in regulatory strategy at early phase, the returns compound. Regulatory investment at seed or Series A does not just reduce risk, it creates optionality at Series B and beyond. This optionality can include cleaner syndication, stronger strategic interest, and faster diligence cycles.
I cannot calculate how many conversations I have had which end in founders concluding "we'll hire regulatory when we need it.” Asserting resources are better spent on continued R&D and attracting additional investors. It is not an assertion that I, a regulatory professional, take as an offense. It is a posture that is increasingly becoming a strategic miscalculation – not a resource decision. A miscalculation that, in the eyes of sophisticated early-stage investors, is made when device design decisions are the most consequential, and least reversible.
This de-prioritizing of regulatory intelligence and its role in early-stage device development has consequences. While it is not a compliance activity, or the operational team responsible for compiling premarket submission and navigating the often cumbersome eSTAR system. It is active competitive Intelligence - ongoing monitoring of FDA trends, competitor authorization, predicate landscape shifts, enforcement actions, and evolving guidance – and the ability to meaningfully leverage this information into value-added advancement.
For late-stage companies, miscalculated failures in regulatory intelligence are well-documented and expensive. Missteps such as a missed predicate withdrawal, or an unanticipated IDE requirement, can imperil a critical Series C/D raises – or even derail strategic exits.
Regardless of development phase, investment in regulatory intelligence and strategy is necessary for short- and long-term success. Companies that treat regulatory intelligence as a standing leadership-level function, not a consultant engagement triggered by a submission, are structurally better positioned across every dimension that investors, acquirers, and regulators evaluate.
What Good Looks Like — A Framework for Executives and Investors
Investing in regulatory is not a one-size-fits-all approach. Experienced executives and investors understand that developing a regulatory intelligence apparatus is not a binary decision – whether that decision is to build, a fractional engagement, or to outsource. The right answer depends on stage, pathway complexity, and proximity to submission — but the decision itself should be made deliberately, not by default. That decision should be made with intent of adapting in the long term. Engaging a consultant to help build your regulatory intelligence operations does not negate developing your own team as resources allow.
For investors, the maturity of regulatory strategy should be consciously scored – not assumed as truth. The presence of a regulatory consultant in cap table discussions can be a positive sign of a startup’s investment in intelligence and strategy – but should not be interpreted as equal to organizational regulatory intelligence. In this capacity, the goal of a consultant is to be a partner in developing the infrastructure of success – passing insights and expertise to clients.
Some key considerations for early-stage startup founders to ask themselves, prior to engaging with investors of any kind.
- Seed / Pre-Seed: Has the team defined intended use with regulatory precision? Is device classification well-reasoned and defensible? Is there a named regulatory pathway with an honest risk assessment attached?
- Series A: Is there a Q-Sub strategy? Is the QMS being built to scale, not just to satisfy a near-term checklist? Is clinical evidence being designed with both the FDA and payer in mind from day one?
- Series B and beyond: Is regulatory strategy integrated with BD and commercial planning? Is the team actively managing predicate landscape and competitive clearance activity? Is there post-market architecture in place before it is required?
Summary
Medtech has entered a K-shaped regulatory risk economy; where risk toleration increasingly favors large Medtech companies operating with uncertainty embedded into overarching strategy, and daily operations. This new reality disfavors startups, conveying those identical risk considerations to sponsors with less experience, minimal comparative resources, and disproportionate pressures on speed and agility. The gap between companies treating regulatory as infrastructure, and those treating it as overhead, is widening and becoming increasingly permanent. In seeking sustainable commercial, and clinical, success – startups must be on the infrastructure side of that divide.
For early-stage founders, the window to build this capability into your company's foundation is moving further upstream, with shorter runways and smaller margins for error than estimated. For investors, the diligence frameworks built and tested in a more forgiving regulatory and capital environment are not sufficient for this one. Regulatory strategy maturity deserves a dedicated evaluation lens, and dedicated capital to further develop.
The leading medical device startups of the next decade will not just have superior technology, and standard of care defining clinical impacts. They will have built regulatory sophistication into their organizational DNA from day one and possess the operational excellence to utilize it as both an offensive and defensive competitive advantage. For these companies, regulatory strategy and intelligence will be a foundational cornerstone. Their outcomes will reflect it, as will their valuations.
1 Market Pathways - What’s Happening at CDRH? RIFs, Retirements, User Fees, and Unknowns
2 Figures intend to convey general concepts. Scenarios depicted are qualitative, not quantitative, and not indicative of response to specific stimuli.